無塵室
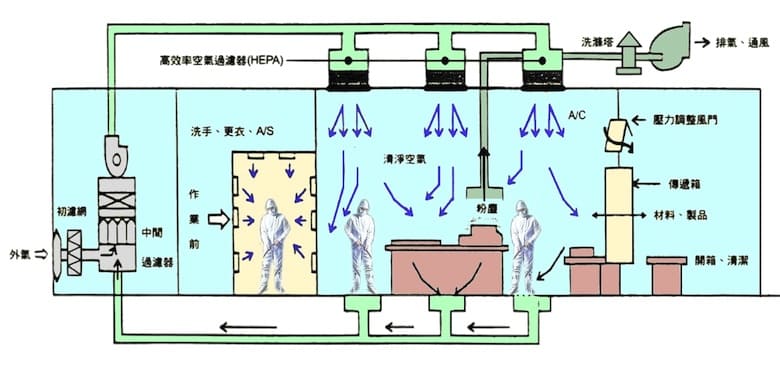
無塵室(或稱:潔淨室)是指一個具有低污染水準的環境,透過將密閉空間內空氣中的微塵粒子等污染物排除,而得到一個相當潔淨的環境。空氣中的污染物包括灰塵,微生物,懸浮顆粒,和化學揮發性氣體。無塵室被廣泛地應用在對環境污染特別敏感的產業,例如半導體生產、生化技術、精密機械、製藥、醫院、無菌食品加工製造業等,其中以半導體業對室內之溫濕度、潔淨度要求尤其嚴格,必須控制在某一個需求範圍內才不會對製程產生影響。
設計無塵室時,應對於生產產品的種類作調查分析,並依據結果進行規劃。包括了解無塵室內設備佈置的規劃、製造的流程、潔淨度之設定、氣流/人流/物流/廢棄物流/製程流之動線、外部環境問題、建築內裝材料之選用等作探討設計。衍生的廣義環境工程包含了溫濕度的控制要求、正負壓、氣流分佈、靜電處理、廢排氣、集塵、二次管配、中央監控系統、空壓及純水處理等。
基本設計原則
- 塵埃之堆積防止
- 塵埃之發生防止
- 塵埃之排除
- 塵埃之不攜入
- 除濕之空調
- 溫濕度控制及壓力之維持
完整規劃之其他考量因素
- 有害氣體之排除
- 結構物與隔間之氣密性
- 靜電之防制
- 電磁干擾之防制
- 備用安全因素之考慮
- 節能之考量
- 預算之考量
- 未來維護及擴建之方便性
無塵室級別標準
無塵室經常使用美國聯邦S209E標準及ISO 14644-1標準所規範的每立方英呎或每立方米最高可容許粒子數目來決定潔淨度級數。
| 級數 | 各大小微塵粒子最高容許量 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0.1 µm | 0.2 µm | 0.3 µm | 0.5 µm | 1 µm | 5 µm | ||||||||
| US F. S209E | ISO 14644-1 | (m³) | (ft³) | (m³) | (ft³) | (m³) | (ft³) | (m³) | (ft³) | (m³) | (ft³) | (m³) | (ft³) |
| ISO 1 | 10 | 2 | 0 | 0 | 0 | 0 | |||||||
| ISO 2 | 100 | 24 | 10 | 4 | 0 | 0 | |||||||
| C-1 | ISO 3 | 1,000 | 35 | 237 | 8 | 102 | 3 | 35 | 1 | 8 | 0 | 0 | 0 |
| C-10 | ISO 4 | 10,000 | 350 | 2,370 | 75 | 1,020 | 30 | 352 | 10 | 83 | 2 | 0 | 0 |
| C-100 | ISO 5 | 100,000 | 3,500 | 23,700 | 750 | 10,200 | 300 | 3,520 | 100 | 832 | 24 | 29 | 0 |
| C-1K | ISO 6 | 1,000,000 | 35,000 | 237,000 | 7,500 | 102,000 | 3,000 | 35,200 | 1,000 | 8,320 | 236 | 293 | 7 |
| C-10K | ISO 7 | * | * | * | * | * | * | 352,000 | 10,000 | 83,200 | 2,360 | 2,930 | 70 |
| C-100K | ISO 8 | * | * | * | * | * | * | 3,520,000 | 100,000 | 832,000 | 23,600 | 29,300 | 700 |
無塵室空調氣流
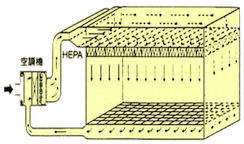
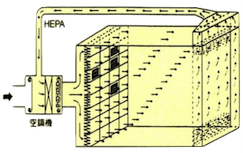
無塵室內的空氣流動方式可分為層流及亂流,其中層流又可再分為垂直層流及水平層流。以下為各種無塵室空調氣流方式的說明及比較。
| 垂直層流 | 水平層流 | 亂流 | |
|---|---|---|---|
| 氣流流線及 設備配置圖 |
 |
 |
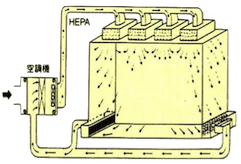 |
| 空氣吹出口 | 佔天花板面積80%~100% | 佔壁板面積80%以上 | 於天花板上使用過濾器吹出口 |
| 空氣吸入口 | 佔地板面積40%以上 | 佔壁板面積40%以上,部分亦可從天花板吸入 | 從地板周圍附近吸入 |
| 適用級數 | Class 1~100 | Class 1~10,000 | Class 1,000~100,000 |
| 換氣次數 | 300~600次/hr | C-100:250~350次/hr C-1K:60~120次/hr |
C-1K:60~120次/hr C-10K:35~45次/hr |
| 優點 |
|
|
|
| 缺點 |
|
|
|
| 價格比 | 85~100 | 40~80 | 20~50 |
食品 TQF 潔淨廠房
一直以來,食品GMP和ISO 22000被視為食品認證的重要指標。食品GMP是良好作業規範(food Good Manufacturing Practices)的簡稱,是一種特別注重製造過程中產品品質與衛生安全的管理制度;ISO 22000食品安全管理系統,整合了ISO 9001品質管理系統和HACCP危害分析重要管制點兩項標準,是國際間重要的食品安全標準。2015年經濟部工業局把GMP標章移轉至民間「台灣優良食品發展協會」(Taiwan Quality Food,簡稱TQF)辦理,加強原物料來源管理及全廠同類產品認證,更加提升了食品供應鏈之安全與品質效益。
隨著大眾對於食品衛生安全的日益重視,為了保障食品在製造過程中不受到環境空氣的污染影響品質,透過無塵室空調系統建立一個具潔淨空氣之作業環境,是經常被使用的方式。因此食品產業鏈上的相關業者,凡舉食品製造、加工、包裝,甚至是物流、倉儲、販賣業者等等,都對於建立能符合食品GMP、ISO 22000、TQF潔淨作業之無塵無菌廠房有相當需求。
下表為美國太空總署NASA NHB-5340.2所定義的無菌室級別,除了微塵粒子之外,同時也規範了微生物的污染上限。NHB-5340.2中微塵粒子的最大容許量與美國聯邦S209E相同級數之容許量相同:
| 無菌室級別 | 微塵粒子的最大容許量 | 微生物污染限量 | ||
|---|---|---|---|---|
| 0.5 µm (個/ft³) | 5 µm (個/ft³) | 懸浮量 (個/ft²) | 沈降量 (個/ft²•週) | |
| 100 | 100 | 0 | 0.1 | 1,200 |
| 10,000 | 10,000 | 65 | 0.5 | 6,000 |
| 100,000 | 100,000 | 700 | 2.5 | 30,000 |
各種食品製造工程之潔淨度需求
| 種類 | 內容 | 適用級別 |
|---|---|---|
| 魚肉加工 | 魚漿冷卻室/包裝室 | 1,000/10,000 |
| 食肉加工 | 漢堡冷卻室/加工、包裝 | 1,000/10,000 |
| 蔬果加工 | 殺菁、冷卻、包裝 | 10,000~100,000 |
| 香菇工廠 | 植苗室/菌室 | 100/1,000 |
| 飲料工廠 | 果汁、鮮奶填充室 | 1,000 |
| 果漿工廠 | PASTE、填充室 | 10,000 |
| 糕餅、製麵 | 冷卻包裝室 | 1,000~10,000 |
| 速食、便當 | 包裝室 (二次加工) | 10,000~100,000 |
食品加工各程序之潔淨度需求
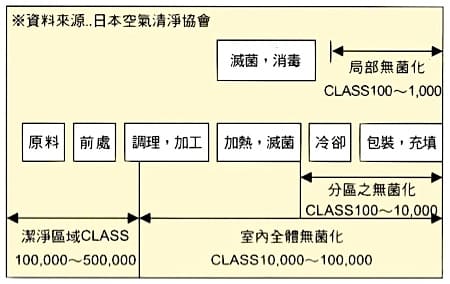
PIC/S GMP 潔淨廠房
隨著GMP的發展,國際間實施了藥品GMP認證。GMP提供了藥品生產和品質管制的基本準則,藥品生產必須符合GMP的要求,藥品品質必須符合法定標準。藥品GMP制度自推行以來,從最初的GMP到cGMP(現行良好作業規範,current Good Manufacturing Practices),至2007年公告實施的國際GMP標準(Pharmaceutical Inspection Convention and Pharmaceutical Inspection Co-operation Scheme,PIC/S GMP,即歐盟GMP標準),不斷推升國內製藥之品質及水準。國產西藥原料藥已於2015年底全面實施GMP(來源)。
藥廠及實驗室為了維持製程或研發環境內空氣之潔淨度,經常使用無塵無菌室控制空氣品質。PIC/S GMP中將無菌製藥的潔淨作業環境分為A、B、C、D四個等級,A級為高風險作業的局部區域,例如,充填區、橡皮塞貯盆、開口安瓿、小瓶及執行無菌連接等區域,通常是以層流工作站的形式建立,B級區為A級區的背景環境,C級與D級區屬於執行較非關鍵性階段的潔淨區。各級環境所需之潔淨度標準如下:
| 等級 | 每立方公尺等於或大於下述粒徑之微粒的最大容許量 | 微生物污染的建議平均限量 | ||||||
|---|---|---|---|---|---|---|---|---|
| 靜態 | 動態 | 空氣樣品 | 落菌培養皿 (直徑90mm) |
接觸培養皿 (直徑55mm) |
手套指印 | |||
| 0.5 µm | 5 µm | 0.5 µm | 5 µm | cfu/m³ | cfu/4hr | cfu/培養皿 | cfu/手套 | |
| A | 3,520 | 20 | 3,520 | 20 | <1 | <1 | <1 | <1 |
| B | 3,520 | 29 | 352,000 | 2,900 | 10 | 5 | 5 | 5 |
| C | 352,000 | 2,900 | 3,520,000 | 29,000 | 100 | 50 | 25 | -- |
| D | 3,520,000 | 29,000 | * | * | 200 | 100 | 50 | -- |
無塵室檢測及確效作業
建置一個無塵室環境,從設計、安裝、檢測、運作及維護,各階段都需要仔細規劃並謹慎地執行,才能達到所期望之目標。為了確保無塵室系統在製程中能保持空氣系統之恆定性,相關檢測及確效作業是不可或缺的。潔淨室確效在製藥方面尤其重要,我國藥品優良製造確效作業基準第四章對空氣系統確效有完整之規定,其中說明確效空氣系統的目的在於確定該系統能持續穩定地控制作業場所之空氣品質,使製成之藥品能維持既訂品質且不受污染。
檢測及確效時機
無塵室進行檢測的時機有三,分別為完工時、備用中、及操作中。並非每一種檢測項目都需要在三個時間點進行測試,而是根據其種類及設計需求而定。
- 完工測試
指無塵室及所有相關支援設施已經完成且可操作,但製程設備及人員均未進場時。 - 備用中(靜態)測試
指無塵室已完成、製程設備已安裝完畢且可操作,但人員尚未進場時。 - 操作中(動態)測試
指無塵室、製程設備、所有相關支援設施及人員已進場且正常運作時。
檢測及確效項目
依據所設計的無塵室系統規格,檢測項目將可能包含下列各項。由於無塵室在驗收基準上有相當之複雜度,須輔以各式精密之量測儀器始可達成。
- 風速、風量及均勻性測試
- 氣流平行性測試
- 懸浮微粒計數測試
- 落菌計數測試
- 溫溼度測試
- 房間壓差測試
- 無塵區隔完整性測試
- 濾網洩漏測試
- 回復測試
- 噪音測試
- 震動測試
- 照度測試







無塵室工程暨確效作業流程
對於製藥產業、侵入性醫療器材產業甚至實驗室而言,產品的品質至關重要,微小的不一致可能帶來嚴重的後果。設計驗證(DQ)、安裝驗證(IQ)、操作驗證(OQ)和性能驗證(PQ),是執行空調系統品質確效的重要方法。下方為無塵室工程暨確效作業之流程及說明。
- 主確效計畫 Master Validation Plan (MVP)
說明哪些系統及設備需要確效、如何確效、及何時進行確效。 - 使用者需求規格 User Requirements Spec. (URS)
使用者對系統之需求規格書,包含目的、範圍、權責及功用。 - 功能規格 Functional Spec. (FS)
具體描述系統及設備可評估的功能或行為之規格書。 - 設計規格 Design Spec. (DS)
提供完整且足夠的規格資訊,用於後續建造系統。 - 設計驗證 Design Qualification (DQ)
確認所提出之設計可滿足 URS 所列要求。 - 安裝驗證 Installation Qualification (IQ) 驗證DS
確認設備於既訂條件下安裝,常用文件包含重要組件查對表、試驗報告、竣工圖、操作維修手冊...等。 - 操作驗證 Operational Qualification (OQ) 驗證FS
測試單一設備或組件在正常及極限範圍下是否能適當運轉。 - 性能驗證 Performance Qualification (PQ) 驗證URS
確認整體系統或設備是否具有穩定表現之性能測試。 - 變更管制 Change Control
凡對系統、設備、程序所作之變更,皆需提供數據或再次確效以保證產品品質。

無塵室工程
工程案例照片


































