洁净室
洁净室(或称:无尘室、无尘车间、净化车间)是指一个具有低污染水平的环境,透过将密闭空间内空气中的微尘粒子等污染物排除,而得到一个相当洁净的环境。空气中的污染物包括灰尘,微生物,悬浮颗粒,和化学挥发性气体。洁净室被广泛地应用在对环境污染特别敏感的产业,例如半导体生产、生化技术、精密机械、制药、医院、无菌食品加工制造业等,其中以半导体业对室内之温湿度、洁净度要求尤其严格,必须控制在某一个需求范围内才不会对制程产生影响。
设计洁净室时,应对于生产产品的种类作调查分析,并依据结果进行规划。包括了解洁净室内设备布置的规划、制造的流程、洁净度之设定、气流/人流/物流/废弃物流/制程流之动线、外部环境问题、建筑装修材料之选用等作探讨设计。衍生的广义环境工程包含了温湿度的控制要求、正负压、气流分布、静电处理、废排气、集尘、二次管配、中央监控系统、空压及纯水处理等。
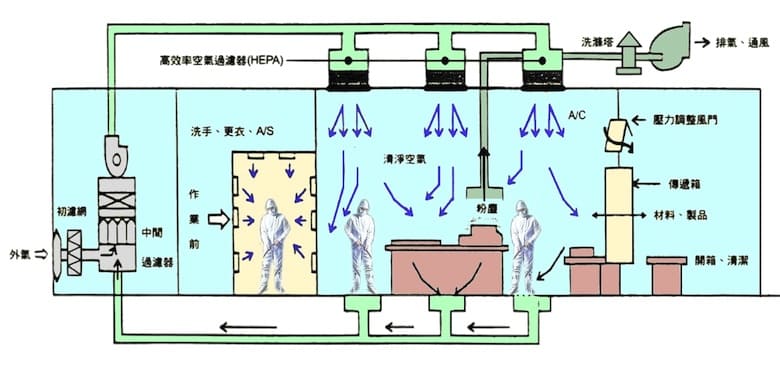
基本设计原则
- 尘埃之堆积防止
- 尘埃之发生防止
- 尘埃之排除
- 尘埃之不携入
- 除湿之空调
- 温湿度控制及压力之维持
完整规划之其他考虑因素
- 有害气体之排除
- 结构物与隔间之气密性
- 静电之防制
- 电磁干扰之防制
- 备用安全因素之考虑
- 节能之考虑
- 预算之考虑
- 未来维护及扩建之方便性
洁净室级别标准
洁净室经常使用美国联邦S209E标准及ISO 14644-1标准所规范的每立方英呎或每立方米最高可容许粒子数目来决定洁净度级数。
| 级数 | 各大小微尘粒子最高容许量 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0.1 µm | 0.2 µm | 0.3 µm | 0.5 µm | 1 µm | 5 µm | ||||||||
| US F. S209E | ISO 14644-1 | (m³) | (ft³) | (m³) | (ft³) | (m³) | (ft³) | (m³) | (ft³) | (m³) | (ft³) | (m³) | (ft³) |
| ISO 1 | 10 | 2 | 0 | 0 | 0 | 0 | |||||||
| ISO 2 | 100 | 24 | 10 | 4 | 0 | 0 | |||||||
| C-1 | ISO 3 | 1,000 | 35 | 237 | 8 | 102 | 3 | 35 | 1 | 8 | 0 | 0 | 0 |
| C-10 | ISO 4 | 10,000 | 350 | 2,370 | 75 | 1,020 | 30 | 352 | 10 | 83 | 2 | 0 | 0 |
| C-100 | ISO 5 | 100,000 | 3,500 | 23,700 | 750 | 10,200 | 300 | 3,520 | 100 | 832 | 24 | 29 | 0 |
| C-1K | ISO 6 | 1,000,000 | 35,000 | 237,000 | 7,500 | 102,000 | 3,000 | 35,200 | 1,000 | 8,320 | 236 | 293 | 7 |
| C-10K | ISO 7 | * | * | * | * | * | * | 352,000 | 10,000 | 83,200 | 2,360 | 2,930 | 70 |
| C-100K | ISO 8 | * | * | * | * | * | * | 3,520,000 | 100,000 | 832,000 | 23,600 | 29,300 | 700 |
洁净室空调气流
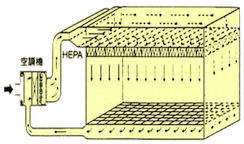
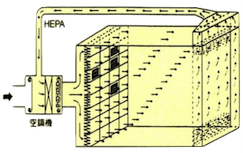
洁净室内的空气流动方式可分为层流及乱流,其中层流又可再分为垂直层流及水平层流。以下为各种洁净室空调气流方式的说明及比较。
| 垂直层流 | 水平层流 | 乱流 | |
|---|---|---|---|
| 气流流线及 设备配置图 |
 |
 |
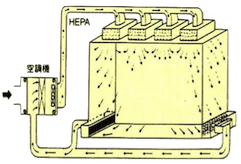 |
| 空气吹出口 | 占天花板面积80%~100% | 占壁板面积80%以上 | 于天花板上使用过滤器吹出口 |
| 空气吸入口 | 占地板面积40%以上 | 占壁板面积40%以上,部分亦可从天花板吸入 | 从地板周围附近吸入 |
| 适用级数 | Class 1~100 | Class 1~10,000 | Class 1,000~100,000 |
| 换气次数 | 300~600次/hr | C-100:250~350次/hr C-1K:60~120次/hr |
C-1K:60~120次/hr C-10K:35~45次/hr |
| 优点 |
|
|
|
| 缺点 |
|
|
|
| 价格比 | 85~100 | 40~80 | 20~50 |
食品 GMP 洁净厂房
食品GMP是良好作业规范(food Good Manufacturing Practices)的简称,是一种特别注重制造过程中产品质量与卫生安全的管理制度;HACCP是危害分析重要管制点(Hazard Analysis and Critical Control Point)的缩写,是一套全球通用的食品安全管理制度;于2005年公布的ISO 22000食品安全管理系统,整合了ISO 9001质量管理系统和HACCP两项国际标准,更加提升了全球食品供应链之安全与质量效益。
随着大众对于食品卫生安全的日益重视,食品GMP和ISO 22000已经成为食品认证的重要指标,为了保障食品在制造过程中不受到环境空气的污染影响质量,透过洁净室空调系统建立一个具洁净空气之作业环境,是经常被使用的方式。因此食品产业链上的相关业者,凡举食品制造、加工、包装,甚至是物流、仓储、贩卖业者等等,都对于建立能符合食品GMP和ISO 22000洁净作业之无尘无菌厂房有相当需求。
下表为美国太空总署NASA NHB-5340.2所定义的无菌室级别,除了微尘粒子之外,另外也规范了微生物的污染上限。NHB-5340.2中微尘粒子的最大容许量与美国联邦S209E相同级数之容许量相同:
| 无菌室级别 | 微尘粒子的最大容许量 | 微生物污染限量 | ||
|---|---|---|---|---|
| 0.5 µm (个/ft³) | 5 µm (个/ft³) | 悬浮量 (个/ft²) | 沈降量 (个/ft²•周) | |
| 100 | 100 | 0 | 0.1 | 1,200 |
| 10,000 | 10,000 | 65 | 0.5 | 6,000 |
| 100,000 | 100,000 | 700 | 2.5 | 30,000 |
各种食品制造工程之洁净度需求
| 种类 | 内容 | 适用级别 |
|---|---|---|
| 鱼肉加工 | 鱼浆冷却室/包装室 | 1,000/10,000 |
| 食肉加工 | 汉堡冷却室/加工、包装 | 1,000/10,000 |
| 蔬果加工 | 杀菁、冷却、包装 | 10,000~100,000 |
| 香菇工厂 | 植苗室/菌室 | 100/1,000 |
| 饮料工厂 | 果汁、鲜奶填充室 | 1,000 |
| 果浆工厂 | PASTE、填充室 | 10,000 |
| 糕饼、制面 | 冷却包装室 | 1,000~10,000 |
| 快餐、便当 | 包装室 (二次加工) | 10,000~100,000 |
食品加工各程序之洁净度需求
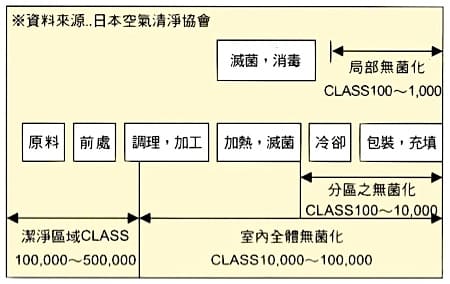
药品 GMP 洁净厂房
随着GMP的发展,国际间实施了药品GMP认证。GMP提供了药品生产和质量管理的基本准则,药品生产必须符合GMP的要求,药品质量必须符合法定标准。中国卫生部亦于1995年颁布《药品生产质量管理规范》,以提升国内人用药之制药质量。2002年,农业部更发布了《兽药生产质量管理规范》(简称兽药GMP),并且于自2006年起强制实施。
药厂及实验室为了维持制程或研发环境内空气之洁净度,经常使用无尘无菌室控制空气质量。国际欧盟 GMP 标准 (PIC/S GMP,Pharmaceutical Inspection Convention and Pharmaceutical Inspection Co-operation Scheme) 将无菌制药的洁净作业环境分为A、B、C、D四个等级,A级为高风险作业的局部区域,例如,充填区、橡皮塞贮盆、开口安瓿、小瓶及执行无菌連接等区域,通常是以层流工作站的形式建立,B级区为A级区的背景环境,C级与D级区属于执行较非关键性阶段的洁净区。各级环境所需之洁净度标准如下:
| 等级 | 每立方公尺等于或大于下述粒径之微粒的最大容许量 | 微生物污染的建议平均限量 | ||||||
|---|---|---|---|---|---|---|---|---|
| 静态 | 动态 | 空气样品 | 落菌培养皿 (直径90mm) |
接触培养皿 (直径55mm) |
手套指印 | |||
| 0.5 µm | 5 µm | 0.5 µm | 5 µm | cfu/m³ | cfu/4hr | cfu/培养皿 | cfu/手套 | |
| A | 3,520 | 20 | 3,520 | 20 | <1 | <1 | <1 | <1 |
| B | 3,520 | 29 | 352,000 | 2,900 | 10 | 5 | 5 | 5 |
| C | 352,000 | 2,900 | 3,520,000 | 29,000 | 100 | 50 | 25 | -- |
| D | 3,520,000 | 29,000 | * | * | 200 | 100 | 50 | -- |
洁净室检测及确效作业
建置一个洁净室环境,从设计、安装、检测、运作及维护,各阶段都需要仔细规划并谨慎地执行,才能达到所期望之目标。为了确保净化空调系统在生产制程中能保持空气系统之恒定性,相关检测及确效作业是不可或缺的。洁净室确效在制药方面特别重要:确效空气系统的目的在于确定该系统能持续稳定地控制作业场所之空气质量,使制成之药品能维持既订高质量且不受污染。
检测及确效时机
洁净室进行检测的时机有三,分别为完工时、备用中、及操作中。并非每一种检测项目都需要在三个时间点进行测试,而是根据其种类及设计需求而定。
- 完工测试
指洁净室及所有相关支持设施已经完成且可操作,但生产设备及人员均未进场时。 - 备用中(静态)测试
指洁净室已完成、生产设备已安装完毕且可操作,但人员尚未进场时。 - 操作中(动态)测试
指洁净室、生产设备、所有相关支持设施及人员已进场且正常运作时。
检测及确效项目
依据所设计的洁净室系统规格,检测项目将可能包含下列各项。由于洁净室在验收基准上有相当之复杂度,须辅以各式精密之量测仪器始可达成。
- 风速、风量及均匀性测试
- 气流平行性测试
- 悬浮微粒计数测试
- 落菌计数测试
- 温湿度测试
- 房间压差测试
- 无尘区隔完整性测试
- 滤网泄漏测试
- 回复测试
- 噪音测试
- 震动测试
- 照度测试







净化工程暨确效作业流程
对于制药产业、侵入性医疗器材产业甚至实验室而言,产品的质量至关重要,微小的不一致可能带来严重的后果。设计验证(DQ)、安装验证(IQ)、操作验证(OQ)和性能验证(PQ),是执行空调系统质量确效的重要方法。下方为净化工程暨确效作业之流程及说明。
- 主确效计划 Master Validation Plan (MVP)
說明哪些系统及设备需要确效、如何确效、及何时进行确效。 - 用户需求规格 User Requirements Spec. (URS)
用户对系统之需求规格书,包含目的、范围、权责及功用。 - 功能规格 Functional Spec. (FS)
具体描述系统及设备可评估的功能或行为之规格书。 - 设计规格 Design Spec. (DS)
提供完整且足够的规格信息,用于后续建造系统。 - 设计验证 Design Qualification (DQ)
确认所提出之设计可满足 URS 所列要求。 - 安装验证 Installation Qualification (IQ) 验证DS
确认设备于既订条件下安装,常用文件包含重要组件查对表、试验报告、竣工图、操作维修手册...等。 - 操作验证 Operational Qualification (OQ) 验证FS
测试单一设备或组件在正常及极限范围下是否能适当运转。 - 性能验证 Performance Qualification (PQ) 验证URS
确认整体系统或设备是否具有稳定表现之性能测试。 - 变更管制 Change Control
凡对系统、设备、程序所作之变更,皆需提供数据或再次确效以保证产品质量。
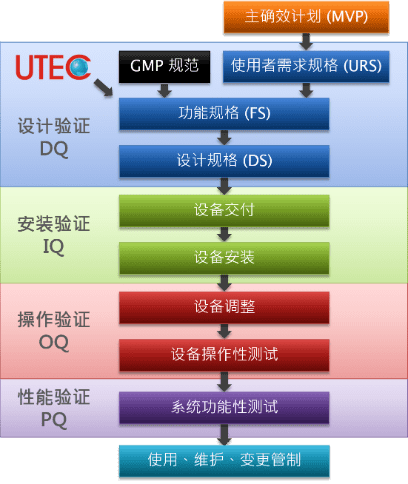
洁净室净化工程
工程案例照片


































